Bio:
- My name is Kevin Legarreta González. I'm studying Computer Sciences in the University of Puerto Rico Rio Piedras campus. My goals are to graduate and be accepted in Boston University to complete a masters in Computer Sciences and Specialize in Cybersecurity or Digital Forensics.
Humor of the week:

Contact info:
- e-mail - kevinleglez@gmail.com
- Github - https://github.com/Kevinlega
Reasearch goals:
-
Fall 2017: Use Mutual technique to implement a good de bruijn graph to do deferential expression.
-
Spring 2017: With titus new comparison code, I will develop a package that will normalize, trim, and compare any given data.
-
Fall 2016: Angel and I intend to implement the transcriptome comparison, that Mutual does, using Khmer Tools. To make it have a faster run time.
Weekly Update:
Fall 2017:
Week 9-14(15/January-20/February):
- Fixed the code so that the memory usage isn't increasing exponentially.
- Both dbg.py and Parser.py don't use GFAPY anymore.
- The parser eliminates links below coverage and segments with no links.
- Tested the parser and with a 2.1 gb output file from dbg.py used 1.79 gb of RAM.
- Have been working on the technical report.
Week 8(7-14/January):
- Added differential expresion to the dbg.py on file named dbgDif.py.
- Also seperated the Differential Expression and the graph creater in two files.
- We can find the graph creator at dbg.py and the DE at Second_program.py
- What we do is:
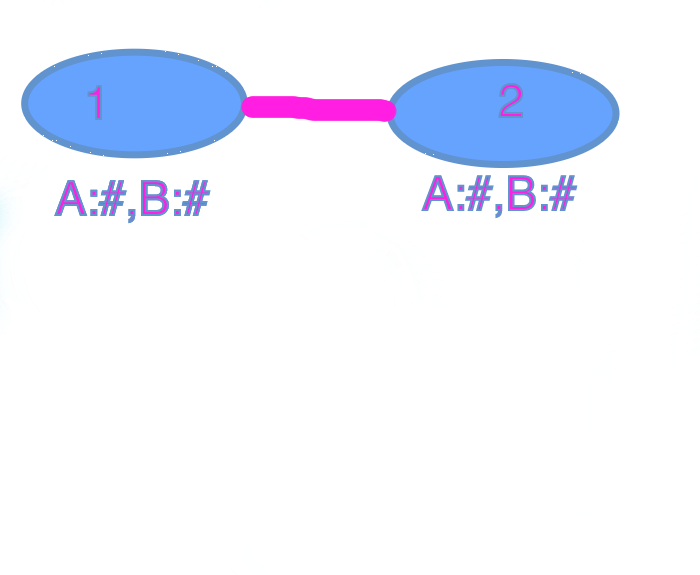
- Take the # from each node and substract them, and add the absolute value of both nodes.
To check if it meets the Threshold (coverage):
if |node1.A# - node1.B#| + |node2.A# - node2.B#| < threshold
delete link
else
keep it
- While at the lab meeting Israel talked about that our approach to splitting the contig is bad because we are duplicating the data in size.
- My next step is to try to send every Kmer count as a comment in GFA1, instead of kmer; also send the complete contig, and the original links. With a parser decide what to remove and what not to remove without splitting into millions of kmers.
Week 5,6,7(December/18/2017-January/5/2018):
- On these weeks I've been working on fixing the dbg.py to detect the kmers to be a kmer as a whole.
- After trying to change the code by our fellow provider, decided to change the tactic and finally have a belived working copy.
- I have the believed working copy here: https://github.com/Kevinlega/DBGDE
-
I did try it between the same file and we have this image from bandage:

-
Next week will finally see my instructor and get the thumbs up; will also talk moving foward.
Week 4(11-15/December):
- Will implement on the dbg.py correct way to take one or more files for two organism.
- Implemented the GFA with the fragments under the segment. On Israel github, we have a working implementation of this: https://github.com/Omig12/Mutual-pepino
- I impemented the write to file for the dbg.py, but later change it to use GFApy library that has this implemented in a function.
- Reading on GFApy, to see if it's to much work to re-implement with this library.
- Used bandage to see our test in the new format, it gave us some random error, we dont know why.
- Implemented a argument parser for the program to take one or more file for two organisms. If more may be needed is in the comments on how to do it. Can be seen at: https://github.com/Kevinlega/DBGDE
- Examples on GFA1 and GFA2: https://github.com/medvedevgroup/TwoPaCo/tree/master/example
- Implemented GFApy on the dbg.py, documentation: http://gfapy.readthedocs.io/en/latest/
- Decided that GFA1 was more helpfull. Used GFA1 with GFAPY to get the output to work with bandage.
- Picture of GFA1 with GFApy working implementation with all the kmers of all the contings:

Week 3(4-8/December):
- Verified the DBG.py we will be using with 3 tests. The tests were:
- With the same file
- With an almost identical file
- With a completely different file
- With this conclude that the dbg.py is doing the same graph, and doing them correctly.
- In our lab meeting on Friday we discussed our goal moving forward and our strategy.
- Decreed that we all would read on GFA format by next lab meeting from: https://github.com/GFA-spec/GFA-spec
Week 2(28/November-1/December):
- First actual lab meeting.
- We decided to use dbg.py taken from: https://github.com/pmelsted/dbg/blob/master/dbg.py
Week 1/2(13-24/November):
- Reading papers
Spring 2017:
Week 17(10-16/July):
- Modified the snakefile to be very friendly.
- Made README.md
- Modified the config file, documented it
- Changed rules and recovered from problems with hulk
- On July 16 going to run the Snakefile on a sample from Holothuroidea (Sea cucumber)
- Next week will start with technical and will run comparison on both Holothuroidea and Nemastostella (provided by Angel)
Week 16(3-9/July):
- Nailed the config file!
- Trimmomatic doens't work on .fasta files
- After trying new softwares decided to go with:
- Instead of changing software and convert from .fastq to .fasta I will go from .fasta to .fastq
- Made the changes. Hulk seems to be down.
- The rule all takes the input to be the last output of the last rule.
- On July 10 will run on hulk the new changes on a fasta file alone, then with the same file, and last run the comparison code on the same file.
- This will be the end of the process.
Week 15(26-30/June):
- Making a configuration file. To make the interaction simpler.
- Trying to make the rule all continue with all the rules.
- Going to make a Menu for the rules. This way if you have multiple organism it will handle all of them before comparing.
- Still working on the menu and config file.
Week 14(19-23/June):
- Work out input on all rules.
- tested each rule individually.
- Was a lot of work. Can't seem to find any paper on input in snakemake.
- Started re-reading all my references to see if I miss something.
- And started making quick summaries on them.
- Will start technical report next week with or without input fixed.
Week 13(12-18/June):
- Huelga done back to buisness.
- Put the Snakefile all together.
- Main issue on the file is the input and the output.
- Still don't understand how to handle input.
- Everything is in the file, but can't test it until I understand input.
- I will begin to do the input and output next week.
- At least all the rules are basically done, only need minor adjustments.
- https://github.com/Kevinlega/Pipeline/blob/master/Snakefile
- Was able to work input only on rule all. On the others it didnt enter. It ran it said:
Provided cores: 1
Rules claiming more threads will be scaled down.
Job counts:
count jobs
1 all
1
rule all:
input: OB/Hour1.fastq, OB/Hour2.fastq, OB/Hour3.fastq, OA/Hour1.fastq, OA/Hour2.fastq, OA/Hour3.fastq
1 of 1 steps (100%) done
Week 12(17-21/April):
- Gave up on input and output.
- Still don't know what wrong with that.
- Finally found something usefull on handling RNA-sequences with Snakemake.
- Been reading this article: http://www.annotathon.org/courses/ABD/practical/snakemake/snake_intro.html
- Helped a lot.
- Going to fix my code to what I have learned.
Week 11(10-14/April):
- Been sick, haven't done work.
- Messing around with my snakemake file on my own.
- Finally got it to run.
- Doesn't seem to detect my dataset!
- Trying to convert just one fastq to fasta file, to test it.
- Didin't have much luck with it.
Week 10(3-7/April):
- Been testing the dummy snakefile I have.
- Downloaded a tutorial snakemake files call kids, sandwiches from: https://github.com/leipzig/SandwichesWithSnakemake
- Worked around with it.
- Too simple to actually work.
Week 9(27-31/March):
- Read the snakemake tutorial. Understood almost all.
- Started getting my code to the snakemake. See it at: https://github.com/Kevinlega/Pipeline/blob/master/snakemake.py
- With angel done with his part, I added his code first.
- I'm developing a try.py to see how the snakemake works with input and output.
- It's not finished because that gave me a hard time. Going to try and find another tutorial.
- If can't find any decent tutorial, going to retake the one before.
Week 8(20-24/March):
- Found diginorm problem, was giving it more 60 GB of RAM xD hulk ain't got that power.
- Trying to divise a way to compute how much diginorm needs.
- Trying...
- Trying ..., ..., ..., still, ..., Inserts windows xp loading sound....
- Have a perfect idea on dividing diginorm RAM needed, will discuss it with Israel and Humberto on Thursday.
- Made the converter tested it.
- The file size from fastq to fasta decreases. Don't know why.
- Tested all scripts. They all work.
- Don't know if the decrease is normal still looking.
- Re-reading Snakemake, and start to move what we have in the makefile to Snakemake.
- Israel and I will write the comparison part and difference, but will wait for the other to finish.
- Let's get to work.
- Snakemake tutorial on the way: https://snakemake.readthedocs.io/en/stable/tutorial/tutorial.html###tutorial
Week 7(6-10/March):
- Made all the scripts in my part, to now start to move them to Snakemake.
- Had problems testing diginorm Im stuck here!!!
- Found how to convert files from fatsq to fasta in BioPython.
- Have a dummy-converter.py still doesn't run.
Week 6(27/Febuary - 3/March):
- Tested all the scripts I have so far, they all seem to work.
- Was doing improvements on the scripts to divide fasta from fastq.
- On Thursday I finished dividing the files, but Israel and I decided to convert the fastq files to fasta
- After this I was going to eliminate all the fastq division I had.
- But got Asthmatic Bronchitis. Haven't been able to do much work after Thursday evening.
Week 5(20-25/February):
- Bad week with only one day of class:
- work on the final touches of the Trinity script.
- Talk to Israel thinking of upgrading all scripts to a python script.
- Worked on concatenating file names, did it, had to read from unix book.
Week 4(13-17/February):
- Finished the script
- It allows you to specify the output folder.
- Sums it into a file call GB.txt
- Also added a not so pretty table call dictionary, but will leave it commented for the moment.
- Presented the Lair paper!
- Did a shell script that will let you run the python scripts on all the files.
Week 3(6-10/February):
- Strated developing a script that will count the total of reads inside fasta and fastq files.
- With this we can calculate how much GB are needed to process the data.
- Reading on Biopython.
- Makefile is comming along.
- Given a lot of thought to the script! It seems imposible.
Week 2(30/January-3/Febuary):
- Reading on Makefile
- Working on Different Makefiles trial, non worked.
- Reading and more Reading
- Going to write description on references.
- On my profile I have a repository of the sketch of makefile. Link: https://github.com/Kevinlega/Wechangethenamelater
- Reading this makefile tutorial: www.gnu.org/software/make/manual/make.html###Makefile-Contents
Week 1(23-27/January):
- Doing research on how to develop software packages.
- Waiting on Hulk to grant me access.
- Downloaded Trimmomatic, Sourmash, and Trinity.
- Will upload them to Hulk.
Fall 2016:
Week 18 and 19(5-15/December):
- Work on the technical.
Week 17(28/November- 2/December):
- Think I have a good plan for the next semester.
- Going to do some research and I'll add it to the technical report.
- Going to read to see what I can improve in the technical report.
Week 16 (21-23/November):
- Humberto showed me the paper that Titus publish on searching.
- Well lets face it, he has the best algorithm.
- Now it up to make a plan for next semester.
Week 15 (14-19/November):
- Running Trinity again.
- If it failes I will download a new file.
- Download sourmash again to read the files.
- Test on Abstract Algebra...
Week 14 (7-11/November):
- Continuing reading on the catlas.
- Re-read the Technical Report to see what can be improved.
- Working on the Cache Lab.
- Trinity failed when running.
Week 13 (1-5/November):
- Reading more scripts on Spacegraphcats.
- Lost the Fasta.fa from Hulk.
- Download a file to run Trinity.
- Trying to fully understand the ways of the catlas.
Week 12 (24-28/October):
- Writing the technical report...
- Going to present Mutual paper
- More technical report...
Week 11 (17-21/October):
- Was able to run multiples spacegraphcats scripts.
- Modified a few scripts in spacegraphcats to see which works best.
- Going to use gimme-dbg-nodes.py and gimme-reads.py and see what it gives.
- I beleive this script hunting might be coming to an end.
- If it works will start to run a script that takes Trinity, spacegraphcats and gives the comparison. -More comming up.
Week 10 (10-14/October):
- Fixed a bug in the spacegraphcats search-dbg.py script.
- Try to do a search on a catlas for a contig inside my genome.
- Didn't work. Because I belive the data is damaged.
- Figure out how to do the search in spacegraphcats for 5% of similarity.
- Running Trinity on a pepino file to do the search again and see if it's my fault.
- Going to the hackaton for the first time! Won't be ablearound until monday!
Week 9 (1-8/October):
- Trying to run Trinity on all the data.
- Found a spacegraphcats script that takes multiple De Bruijn Graphs and compares them.
- Can't test the scrip[t yet. Can't find the graph output of Trinity.
- Using all my might to be able to run Trinity by Saturday.
- Searching for the graph to test the spacegraphcats script.
Week 8 (26-30/September):
- Trinity was runned with the same file as Mutual.
- Mutual output twice as much for no reason.
- Reading on the art of khmer tools.
- Trying to understand spacegraphcats.
- Program here we go!
Week 7 (18-23/September):
- Running Mutual. Hope for good results this time.
- Tried to run Trinity, said something about perl.
- Fixed perl problem, but still won't run.
- Will compare Velvet/Oases run time vs Trinity on the same fastq file, but trinity doesn't run.
- Do to the "Blackout, blackout" (see In the Heights musical) couldn't do much work.
- Mutual was running error. Found out that if you don't erase corrupted will never run.
- Also velvet had an update. Found out you should do a git pull once in a while.
- run updatevelvet.sh, new update makefile of oases does make to velvet too.
- Doing the make only on oases won't get velveth and velvetg.
- Well during the blackout, all the parts for my computer came.
- Is up and running, its name is SPIDER....! ubuntu 16.04!
Week 6: (12-17/September)
- Taking Trinity tutorial via a virtualbox.
- Download Trinity, spacegraphscats.
- Reading on khmer tools.
- Reading on Trinity.
- Reading on spacegraphscats.
- The output of Mutual data, was damaged by the crash.
- On Monday morning will start Mutual again, because of the maintenance.
- Bought everthing for a "supercomputer" going to learn how to set it up.
- Also need to build a bootable linux (ubuntu) usb.
- Final, I will learn how to open an ssh.
Week 5: (5-10/September)
- Mutual was running on hulk, but it felt.
- Download Sea cucumbers transcriptomes from NCBI database
- Download FastQC and Trimmomatic.
- Run FastQC in one of the fastq before running Trimmomatic.
- After trimming the data, ran FastQC on the clean data.
- Reading Khmer tools, question have been present in the research.
- Coulnd't make it to meeting, health problems on thursday, lost all night in Hospital.
Week 4: (29/August - 2/September)
- Compiled oases, velvet and mutual on hulk.
- Created a script to run the whole program.
- Continue reading on khmer tools.
- Tried to install blast, I can't.
- Waiting to run Mutual.
Week 3: (22-26/August)
- Created a github project for the investigation (TrinihMer)
- Angel and I worked on uploading the mutual project on hulk to test it.
- Is showing an error, will try to understang this weekend.
- Reading about Khmer Tools.
- Uploaded a clean sequence to the hulk for the test of Mutual.
- We are supposed to start writing code next week.
- Will contact Angel to see when can we meet to work on the program.
- The data used to test Mutual is on the /tmp - hulk
- Excited to start coding the transcriptome comparison code.
- Created a workspace in hulk to have khmer and networkx, that way we will work on hulk.
Week 2: (15-19/August)
- Trying to run Mutual on my computer.
- Download Velvet and Oases. Was able to compile.
- Trying to compile Mutual, doesn't work do to the compiler's version my computer has.
- Re-installed a C++ compiler to see if that is the problem... still installing.
- Had a Reunion with Israel. He and I worked on Mutual, Oases and Velvet until the computer would compile them.
- Will download a Rat sequence to try software.
- Hopefully starting next week, the code will begin to be re-written in a more efficient way!
Week 1: (9-12/August)
- Gained access to megaprobe lab
- Started to research on terms of bioinformatics.
- Read Israel’s Technical Report.
- Read Papers assigned by Humberto Ortiz.